
Research focus
Obesity is increasing exponentially in western society. Similarly, osteoporosis is endemic in our country as half of Asian and White women reaching the age of 65 years will experience a spontaneous fracture. Our laboratory has for many years studied the mechanisms by which osteoclasts degrade bone and as a result, our trainees have become the leaders in the field of bone biology. Because it has become evident that obesity compromises bone health and predisposes to fracture we have expanded our interests to explore the relationship of fat and bone as well as the mechanisms of weight control. Our efforts have established that “fat talks to bone” and that macrophage lineage cells, which differentiate into osteoclasts, also regulate the development of obesity. Thus, while our lab focuses on a number of fat and bone related projects, three are paradigmatic of our efforts:
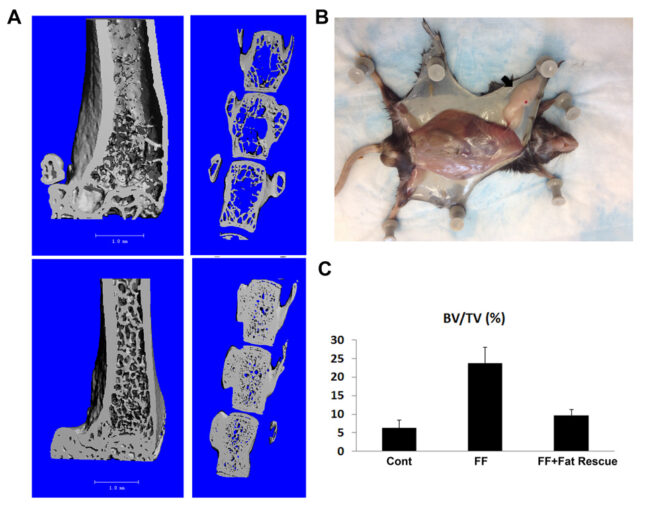
Given the clinical relationship of obesity and the skeleton, we hypothesized that fat-secreted molecules influence skeletal mass by directly and indirectly targeting bone cells. We reasoned that a fat-depleted mouse with a robust bone phenotype, normalized by adipocyte transplantation, would be an exceptional venue to establish how fat talks to bone. This venue would enable determination of the influence of individual fat depots as well as specific adipocyte-produced molecules on the skeleton. It will establish if bone accrual is linearly related to fat abundance or whether a “threshold” of weight loss must be achieved before skeletal mass is enhanced. These studies would also provide information as to whether aging compromises the beneficial effects of weight loss on bone. Understanding the means by which fat, in its various forms, impacts bone, may provide a foundation for preventing and treating the skeletal complications of the metabolic syndrome. To determine if this is so, we generated mice in which fat is germline deleted and another in which fat is ablated post-nataly. In both circumstances, bone mass is increased at least 5 fold but by different mechanisms. Given their dramatic skeletal phenotypes, these fat depleted mice provide a unique venue to determine how distinct forms of fat and their components signal to bone lineage cells to modulate skeletal mass questions of profound clinical implications.
Presently, body weight is largely controlled by restricting food intake. This approach is often unsuccessful because obese patients who lose weight by dieting are unable to sustain their leaner phenotype due to decreased resting energy expenditure. Thus, a strategy which increases energy expenditure without significantly compromising insulin sensitivity may prevent recurrent weight gain. Our data indicate such a strategy is feasible. ASXL2 is a protein expressed conjointly with myeloid differentiation genes. ASXL2-/- mice are insulin resistant and have decreased body fat (lipodystrophy). As expected because of lipodystrophy, ASXL2-/- mice fail to gain weight on a high fat diet which we attributed to deletion of the gene in adipose tissue. Surprisingly, however, high fat diet mice in whom ASXL2 is conditionally deleted only in myeloid cells, completely mirror their ASXL2-/- counterparts in that they completely fail to gain weight despite fatty food intake and activity equivalent to WT mice whose body mass increases 50 % within 10 weeks. Importantly, whereas WT high fat diet mice are insulin insensitive, ASXL2LysM high fat diet mice are not. Furthermore, energy expenditure is increased 43% in ASXL2LysM mice. Our data indicate that the means by which myeloid cells (macrophages) lacking ASXL2 prevents obesity involves their switch from a pro-inflammatory (M1) to a pro-immune (M2) phenotype. We have therefore established that deletion of ASXL2, exclusively in myeloid lineage cells, prevents high fat diet induced obesity by enhancing energy expenditure while maintaining insulin sensitivity. Our goal is to determine the mechanism whereby ASXL2-deficient myeloid cells prevent high fat diet induced obesity and identify a potential anti-obesity target. If successful, our efforts could profoundly impact the health of our society.
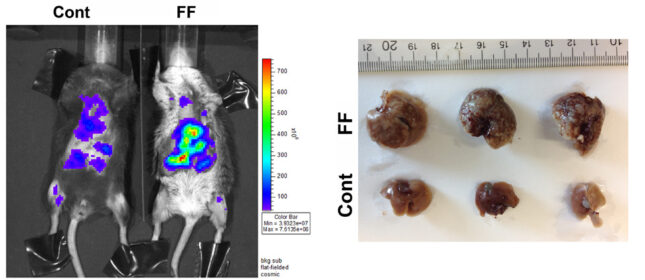
Both fat depletion (lipodystrophy) and obesity cause non-alcoholic fatty liver disease (NAFLD) which is endemic in our society. In our study of fat and bone, we unexpectedly found that NAFLD markedly increases liver metastasis. NAFLD promotes metastatic growth by providing hepatocyte derived fatty acids as an energy source for cancer cells. Evidence indicates this transfer of fatty acids from hepatocytes to cancer also occurs in patients. We are exploring how lipids are transferred from fatty hepatocytes to cancer cells and the role the tumor microenvironment plays in the process. We have shown that reversal of fatty liver prevents metastasis and are determining if anti-NAFLD dugs have an anti-neoplastic effect. If our hypothesis translates to patients, NAFLD prevention becomes essential to all those with potentially metastatic cancer.
Selected Publications:
- Zou W, Teitelbaum SL. Absence of Dap12 and the αvβ3 integrin causes severe osteopetrosis. J Cell Biol. 208(1):125-36, 2015.
(Journal Highlight: Leslie M. No substitute for β3 integrin. J Cell Biol 208(1):2, 2015) - Zou W, Rohatgi N, Brestoff JR, Moley JR, Li Y, Williams JW, Alippe Y, Pan H, Pietka TA, Mbalaviele G, Newberry EP, Davidson NO, Dey A, Shoghi KI, Head RD, Wickline SA, Randolph GJ, Abumrad NA, Teitelbaum SL. Myeloid-specific Asxl2 deletion limits diet-induced obesity by regulating energy expenditure. J Clin Invest. 130(5):2644-2656, 2020.
- Zou W, Rohatgi N, Brestoff JR, Li Y, Barve RA, Tycksen E, Kim Y, Silva MJ, Teitelbaum SL. Ablation of Fat Cells in Adult Mice Induces Massive Bone Gain. Cell Metab. 32(5):801-813.e6, 2020
(Selected as Free Featured Article in Cell Metabolism) - Li Y, Su X, Rohatgi N, Zhang Y, Brestoff JR, Shoghi KI, Xu Y, Semenkovich CF, Harris CA, Peterson LL, Weilbaecher KN, Teitelbaum SL, Zou W. Hepatic lipids promote liver metastasis. JCI Insight. 5(17):e136215, 2020
(Selected as Editor’s Pick in October issue of JCI This Month)
Current projects
While obesity has long been considered beneficial for skeletal health recent studies suggest the opposite. Thus, despite its demographic importance, the influence of fat on bone remains enigmatic. Although controversial, studies of the effect of fat-produced molecules, such as leptin and adiponectin, indicate these selected adipokines impact bone. Adipose tissue is, however, a complex organ and there is little mechanistic insight as to how fat, per se, and which variety of fat, regulates the skeleton.
Such information is clinically relevant as individuals with a predominance of visceral fat, as attends the metabolic syndrome, are osteopenic whereas subcutaneous and brown fat may positively influence bone mass. To address this issue we generated mice completely lacking visceral, subcutaneous and brown fat. Despite the hypogonadal state of these “fat free” (FF) mice, trabecular bone volume is enhanced approximately 400-500% with a significant increase in cortical thickness, due to robust osteoblast proliferation. Unexpectedly in face of its marked increase in bone mass, osteoclasts in FF mice are also markedly enhanced. This observation raises the possibility that the abundance of bone in these animals is the product of accelerated skeletal remodeling initiated by the abundant osteoclasts. Our observations establish that, by mechanisms to be determined, fat signals to bone and decreased adiposity may greatly increase bone mass, challenging the concept that obesity improves skeletal health. Most importantly, the skeletal phenotype of FF mice is completely rescued by adipocyte precursor transplantation. This transplantation-mediated normalization of FF bone provides the opportunity to directly explore the impact of deleting various adipocyte products on bone accrual and how visceral, subcutaneous and/or brown adipose tissue targets the skeleton.
Obese patients who lose weight are unable to sustain their leaner phenotype because of decreased resting energy expenditure. Thus, a strategy which increases energy expenditure without significantly compromising insulin sensitivity may prevent recurrent weight gain. Our data indicate such a strategy is feasible. We have discovered that mice in whom a gene was deleted exclusively in myeloid lineage cells (skinny mouse) surprisingly completely fail to gain weight despite high fat diet (HFD) intake and activity equivalent to WT mice whose body mass increases 50 % within 10 weeks.
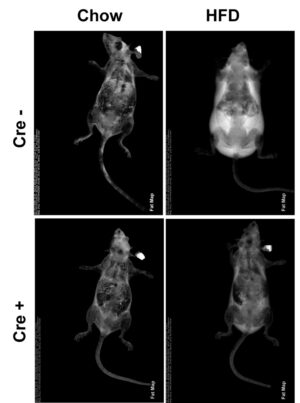
Importantly, whereas WT HFD mice are markedly insulin insensitive, skinny HFD mice are similar to chow fed WT. Furthermore, energy expenditure, as manifest by VO2, is amplified 43% in skinny mice. Fecal fat of HFD skinny and WT mice are indistinguishable establishing the striking obesity resistance does not reflect intestinal malabsorption. We have therefore established that deletion of a gene, exclusively in myeloid lineage cells, prevents HFD-induced obesity while essentially maintaining insulin sensitivity. The goal of this exercise is therefore, to determine the mechanism whereby modification of myeloid cells prevents HFD-induced obesity and identify a potential anti-obesity target.
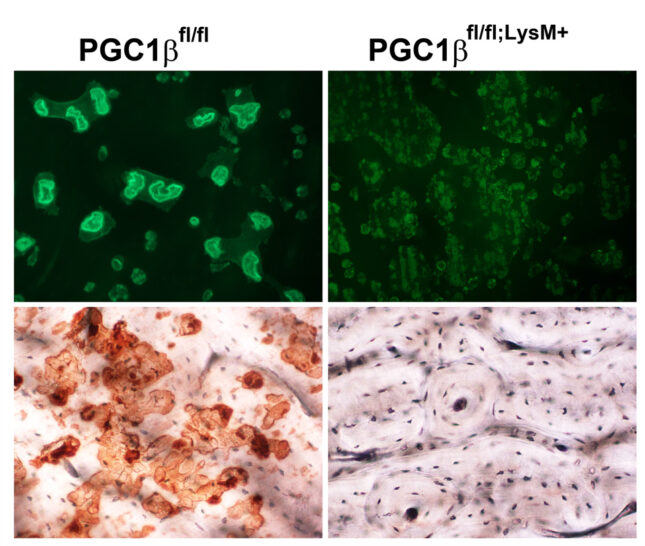
Osteoclasts are the most mitochondria-rich cells in man. Peroxisome proliferator-activated receptor gamma coactivator 1-β (PGC1β) is a transcriptional coactivator that regulates energy metabolism by stimulating mitochondrial biogenesis and respiration. Previous studies using global knockout mice reported that PGC1β is essential for osteoclast differentiation. This conclusion is inconsistent, however, with the normal number of osteoclasts PGC1β-/- mice. To determine the role of PGC1β in osteoclastogenesis, we generated conditional knockout mice, in which PGC1β is exclusively deleted in myeloid lineage cells (PGC1βLysM). Osteoclast maturation was determined as a function of differentiation markers and TRAP staining. Mature osteoclast function was assessed by actin ring and resorptive pit formation on bone. Cytoskeletal organization was monitored by live fluorescent imaging of GFP-labeled actin. Rac1 activation by M-CSF and vitronectin was detected by GTP pull-down assay and western blotting. Mitochondrial biogenesis and mitochondrial complex subunit expression were determined by qPCR and Western blotting, respectively. Mitochondrial number was detected using mitochondrial specific probes. Challenging previous reports, we find osteoclast differentiation of PGC1β-deficient bone marrow macrophages (BMMs), as determined by the unchanged expression level of differentiation markers including β3 integrin, c-Src, NFATc1, and Cathepsin K, is unaltered.
On the other hand, osteoclast function is impaired when PGC1β is deleted, as manifest by a complete absence of actin rings and resorptive pits on bone. Surprisingly, PGC1β-deficient osteoclasts are huge likely due to marked hypermobility thereby increasing contact with mononuclear precursors. Rac expression and Rac1-GTP formation are unaltered in PGC1βLysM osteoclasts establishing a non-canonical mechanism generates their markedly abnormal cytoskeleton. In this regard, mitochondrial biogenesis, mitochondria number, and expression of mitochondrial respiratory chain proteins are significantly decreased in the PGC1β-deficient BMMs and osteoclasts. Our results establish that PGC1β is essential for osteoclast function but not differentiation and indicate that their cytoskeletal organization is mitochondria-mediated. Thus, the role of this exercise is to delineate the role of mitochondria in osteoclast cytoskeletal organization.
Breast cancer is among the most common causes of female death. While major advances have been made in treating this malignancy, once breast cancer has metastasized to liver, approximately 80% of patients will die within 5 years. Thus, insights into circumstances promoting breast cancer metastases to liver, particularly if preventable, would have profound effects on public health. Given evidence that fatty acids promote tumor growth we reasoned that one such preventable possibility is fatty liver disease. Hepatic steatosis, as a product of the metabolic syndrome, is endemic in western society. While many complications of fatty liver have been documented, its effects on metastasis are surprisingly unknown. Interestingly, mice are resistant to hepatic breast cancer metastasis providing the opportunity to explore preventable cellular and molecular events which may predispose to this condition. To this end we systemically administered breast cancer cells to 3 murine models of hepatic steatosis. WT mice exhibited little or no hepatic metastasis while, in each circumstance, steatotic livers were replete with cancer. There were no differences in tumor abundance in other organs. Metastasis to steatotic livers also resulted in a virtual complete removal of hepatocyte fat raising the possibility that cancer growth reflects utilization of fatty acids as fuel. In keeping with the posture that steatotic lipids may promote metastatic growth, PET scanning reveals robust uptake of radiolabeled palmitate by liver-residing breast cancer. Thus, our data establish that hepatic steatosis predisposes to liver metastasis in mice which, if true in patients, has profound public health implications. We hypothesize that the predisposition of breast cancer to metastasize and grow in fatty livers reflects, at least in part, utilization of hepatocyte-residing lipids as fuel. We will therefore determine the metabolic events by which hepatic steatosis predisposes to liver metastasis.
To this end, we will define the rates of breast cancer homing to WT and steatotic livers and its rate of growth once established. To gain mechanistic insights we will explore the means by which breast cancer accumulates hepatocyte-residing fatty acids and the impact of hepatic lipids on tumor energy utilization. We will also address the possibility that circulating factors, attending insulin resistance, contributes to liver tumor growth. These studies will provide a foundation for exploring the possibility that preventing fatty livers in patients, particularly those with adult onset diabetes and obesity, will reduce spread of breast cancer and thus prolong life expectancy.
Publications
Lab members
- Nidhi Rohatgi, PhD, Instructor in Pathology and Immunology
- Wei Zou, MD, PhD, Assistant Professor of Pathology and Immunology
- Yousef Abu-Amer, PhD
- Zvi Bar-Shavit, PhD
- Jonathan Brestoff, MD, PhD, MPH
- Xu Cao, PhD
- Denis Clohisy, MD
- Carl DeSelm, MD, PhD
- Roberta Faccio, PhD
- David Lacey, MD
- Yongjia Li, PhD
- Sunao Takeshita, PhD
- Deborah Veis (previously Novack), MD, PhD
- Julia Warren, MD, PhD
- Yan Zhang, PhD
- Haibo Zhao, MD, PhD
Join this lab

To inquire about available positions for DBBS graduate students or postdocs, please contact Dr. Teitelbaum by email at teitelbs@wustl.edu.